




Entries in genetics (27)

 Adrian Liston
Adrian ListonDecennia-oude, mysterieuze ziekte geïdentificeerd en mogelijke behandeling gevonden
 Een mysterieuze ontstekingsziekte teistert al drie generaties lang een Vlaamse familie met ernstige huidletsels, koorts, pijn en uitputting. De ziekte, waarvoor men tot nu toe geen oorzaak of behandeling had gevonden, is nu geïdentificeerd als pyrine-geassocieerde auto-inflammatie met neutrofiele dermatose (Pyrin Associated Autoinflammation with Neutrophilic Dermatosis, afgekort PAAND), en werd ook vastgesteld bij families in Engeland en Frankrijk. In een nieuw onderzoek hebben Adrian Liston (VIB/KU Leuven) en Carine Wouters (UZ Leuven/KU Leuven) de genetische mutatie ontdekt die de ziekte veroorzaakt, en ook een doeltreffende behandeling gevonden. Hun onderzoek werd gepubliceerd in het internationale wetenschappelijke tijdschrift Science Translational Medicine.
Een mysterieuze ontstekingsziekte teistert al drie generaties lang een Vlaamse familie met ernstige huidletsels, koorts, pijn en uitputting. De ziekte, waarvoor men tot nu toe geen oorzaak of behandeling had gevonden, is nu geïdentificeerd als pyrine-geassocieerde auto-inflammatie met neutrofiele dermatose (Pyrin Associated Autoinflammation with Neutrophilic Dermatosis, afgekort PAAND), en werd ook vastgesteld bij families in Engeland en Frankrijk. In een nieuw onderzoek hebben Adrian Liston (VIB/KU Leuven) en Carine Wouters (UZ Leuven/KU Leuven) de genetische mutatie ontdekt die de ziekte veroorzaakt, en ook een doeltreffende behandeling gevonden. Hun onderzoek werd gepubliceerd in het internationale wetenschappelijke tijdschrift Science Translational Medicine.
Al decennia lang kampen families in België, Engeland en Frankrijk met een mysterieuze ziekte die huidletsels, koorts, pijn en uitputting veroorzaakt. Elke generatie krijgt de helft van de kinderen van personen die de ziekte hebben, dezelfde symptomen. Artsen waren er niet in geslaagd de ziekte te identificeren of een doeltreffende behandeling te vinden. Nu is de identificatie eindelijk een feit en is dankzij een internationaal onderzoeksteam ook een behandeling gevonden.
Prof. Adrian Liston (VIB/KU Leuven, hoofd van het wetenschappelijk onderzoeksteam): “Dankzij het nauwgezette werk van de artsen weten we nu dat we te maken hebben met een erfelijke aandoening. Dankzij de vooruitgang in de DNA-sequentietechnologie konden we het genoom van deze patiënten bepalen en de mutatie opsporen die de ziekte veroorzaakt.”
Het gaat om een mutatie in het MEFV-gen. Mensen die van hun beide ouders een MEFV-gen met een mutatie overgeërfd hebben, lijden aan de ontstekingsziekte familiaire mediterrane koorts (FMF), een ontstekingsziekte. Bij PAAND-patiënten gaat het echter om een andere mutatie in het MEFV-gen én is één enkele kopie van de mutatie voldoende om de ziekte door te geven. Dit betekent dat de helft van de kinderen van de patiënten de ziekte overerven, in tegenstelling tot de mutaties die FMF veroorzaken (die vaak een generatie overslaan). De PAAND-mutatie zorgt ervoor dat het lichaam reageert alsof er een bacteriële huidinfectie plaatsvindt. Daardoor gaat de huid het ontstekingseiwit interleukin-1β produceren, dat huidletsels, koorts en pijn veroorzaakt.
Een behandeling voor de nieuwe ziekte?
Dankzij het opsporen van de biologische oorzaak van deze ziekte kon men ook een nieuwe behandeling bepalen. De onderzoekers hergebruikten anakinra (Kineret ®), een middel tegen artritis dat zich richt tegen interleukin-1β, dat ook bij PAAND een belangrijke rol speelt. De resultaten bij een eerste patiënt, uit een Engels gezin, waren opvallend positief. De huidletsels verdwenen snel en de patiënt herstelde helemaal van de koorts en de pijn. Op dit moment wordt een uitgebreidere test uitgevoerd bij Vlaamse patiënten, om te zien of deze gerichte behandeling tot een volledige genezing kan leiden.
Prof. Carine Wouters (KU Leuven/UZ Leuven, hoofd van het klinische onderzoeksteam): “Dit is het resultaat van een intense samenwerking tussen artsen en wetenschappers die al bijna tien jaar de ziekte trachten te begrijpen. Ik ben verheugd vast te stellen dat we deze zeldzame mutatie nu beter begrijpen en dat we voor deze patiënten de weg hebben geopend naar een doeltreffende therapie.”
Citaat van een patiënt: “We zijn blij en heel dankbaar dat de artsen en wetenschappers hun zoektocht naar de oorzaak van de ziekte die onze familie al zo lang treft, nooit hebben gestaakt. We hopen dat de nieuwe behandeling gunstig zal zijn voor onze familie. En we beseffen ook dat de bevindingen andere patiënten zullen helpen om een correcte diagnose en behandeling te krijgen.”
Prof. Adrian Liston (VIB/KU Leuven, hoofd van het wetenschappelijk onderzoeksteam): “Dit is een uitzonderlijke periode voor het onderzoek rond erfelijke aandoeningen. We helderen elke maand klinische gevallen op die enkele jaren geleden nog niet op te lossen waren. We ontdekken nieuwe mutaties en beschrijven nieuwe ziektebeelden en ziektemechanismen waarvoor ook nieuwe werkzame geneesmiddelen kunnen worden voorgeschreven. Patiënten komen daardoor soms in moeilijke situaties terecht, waarbij de wetenschap een oplossing kan bieden, maar de ziekteverzekeringen de kosten voor geavanceerde diagnosetests of nieuwe behandelingen nog niet kunnen terugbetalen. Dit vormt dan ook een uitdaging voor zowel de farmaceutische industrie als de overheid. Zowel nieuwe medicijnen als bestaande medicijnen voor nieuwe indicaties dienen ter beschikking worden gesteld van patiënten die – op basis van genetische testen – zeer goed kunnen gedefinieerd worden.
Prof. Carine Wouters en prof. Adrian Liston hebben het Leuven Universiteitsfonds Ped IMID (Pediatrische Immuun-inflammatoire aandoeningen) opgericht, een waarmee ze middelen willen werven om onderzoek, diagnose en behandeling mogelijk te maken voor personen die lijden aan zeldzame immuunziekten die momenteel niet door de ziekteverzekeringen worden gedekt.
Ook gelezen: De Staandard, Het Laatste Nieuws, Het Nieuwsblad, De Morgan
Decades-old mystery disease identified and potential cure found
A mysterious inflammatory disease has been afflicting a Flemish family for three generations, causing severe skin lesions, fevers, pain and exhaustion. This disease, which previously had no known cause or cure, has now been identified as Pyrin Associated Autoinflammation with Neutrophilic Dermatosis (PAAND), and has also been found in families in England and France. New research by Adrian Liston (VIB/University of Leuven, Belgium), Seth Masters (Walter and Elisa Hall Institute, Australia), Carine Wouters (University of Leuven, Belgium) has found the genetic mutation causing the disease and also identified an effective treatment. This research was published in the international scientific journal Science Translational Medicine.

For decades, families in Belgium, England, and France have been living with a mysterious disease that results in skin lesions, fevers, pain and exhaustion. Every generation, half of the children of the people with this disease develop the same symptoms. Doctors had been previously unable to identify the disease or find any effective treatment. For the first time, this disease has been identified and a treatment found due to an international research team.
Professor Adrian Liston (VIB/University Leuven): “Detailed work by clinicians told us that we were dealing with a genetic disease. Thanks to advances in DNA sequencing technology we were able to sequence the genome of these patients and find the mutation causing the disease”.
The mutation is in the gene called MEFV. This gene was known to cause an inflammatory disease called Familial Mediterranean Fever (FMF) in patients who inherit mutated copies from both mother and father. However, the mutation found in the PAAND patients is different. Only a single copy of the mutation is needed to cause disease, meaning it affects half the children of patients, unlike the mutations that cause FMF, which often skip generations.
Professor Seth Masters (Walter and Elisa Hall Institute): “The PAAND mutation causes the body to as if there is a bacterial skin infection. This leads to the skin making the inflammatory protein interleukin-1β, which causes skin lesions, fevers and pain”.

A cure for the new disease?
Understanding the biological basis for this new disease allowed the rational selection of a new treatment. The researchers repurposed an anti-arthritis drug, anakinra, which targets the same protein that causes PAAND, interleukin-1β. The results in the first volunteer, from an English family, were striking, with a rapid clearance of skin lesions and a complete recovery from fevers and pain. A larger trial is now beginning in the Flemish patients to see if this targeted treatment will act as a complete cure.
Professor Carine Wouters (KU Leuven/UZ Leuven, lead clinical researcher): “This is the synthesis of an intense collaboration between clinicians and scientists trying to understand this disease for almost 10 years. I am delighted to see how it has increased our understanding of rare mutations, and especially has opened a therapeutic perspective for these patients.”
Quote from one of the patients: “We are happy and very grateful to the doctors and scientists who never gave up their search to understand the disease that affected members of our family for so many years. We are very hopeful that the new treatment will be beneficial to our family. Also we realize that the findings will help other patients to get a correct diagnosis and therapy.”
Professor Adrian Liston (VIB/KU Leuven, lead scientific researcher): “This is an amazing time to be working on genetic diseases. Every month we are solving clinical cases that would have been too hard to work out just a few years ago. Actually, to be honest the research is moving much faster than the healthcare system – we are finding new mutations, new diseases and trialling new treatments faster than the healthcare system is adapting. It creates a difficult situation for patients where the science is in, but the health insurance funds are not ready to reimburse the costs of the advanced diagnostic tests we use or novel treatments that we discover. This is a challenge, but also an opportunity – medical improvements could be rolled out quite quickly with political will.”
Professors Carine Wouters and Adrian Liston have established the charity Ped IMID to seek funding for research, diagnosis, and treatment of people living with rare immune disorders not currently covered by the health insurance funds.
To read more, go to our article in Science Translational Medicine:
Masters, Lagou, Jéru, Baker, Van Eyck, Parry, Lawless, De Nardo, Garcia-Perez, Dagley, Holley, Dooley, Moghaddas, Pasciuto, Jeandel, Sciot, Lyras, Webb, Nicholson, De Somer, van Nieuwenhove, Ruuth-Praz, Copin, Cochet, Medlej-Hashim, Megarbane, Schroder, Savic, Goris, Amselem, Wouters* and Liston*. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Science Translational Medicine. 2016 in press.
Yes, diabetes is a genetic disease
The genetic basis for diabetes has been long established. It really is a shame that diabetes is so often called a "lifestyle disease". It isn't. 50% of the susceptibility to type 1 diabetes is genetic, and 70% of the susceptibility to type 2 diabetes is genetic.
Most of the remainder of the susceptibility is environmental. For example, certain types of dietary fat, such as palmitate, increase the fragility of beta cells. It is quite plausible that much of the increase in diabetes incidence in the past decades is due to changes in diet that make our beta cells more fragile. But even when we are talking about dietary factors, I think it is important to recognise that much of the effect is environmental rather than lifestyle. "Lifestyle" is easy to dismiss by blaming the patient for their own choices. Calling diabetes a lifestyle disease is one of the reasons that research and medical advances in diabetes are lagging behind, and the stigma contributes to the ill effects of diabetes (e.g., increased risk of depression, more antagonistic relationships between patients and clinicians). Recognising diet as an environmental factor takes away this stigma, is more accurate and allows us to tackle the problem using public health approaches. For example, some of the highest rates of diabetes are in the poorest neighbourhoods of America. In these areas, junk food is cheap and available everywhere, while good food is simply not practical - it is rare, expensive and takes more time to prepare than many poor families have. Likewise, the neighbourhoods may not provide the physical infrastructure that allows for a mobile lifestyle (parks for kids, urban design that promotes walking, etc). It is not a "lifestyle choice" to live in these diabetogenic environments, but it is a public policy choice to allow these environments to exist.
To really tackle the diabetes epidemic we need to recognise that the root cause is not in personal choices made by individuals. The root cause is in the social structure that we have created, in the urban design of cities, the changes in food culture, the demands placed on our time. We have made unhealthy lives the easiest to live. We can either ask individuals to make heroic efforts to overcome these obstacles to a healthy lifestyle, or we can use public policy to make our food environment and urban environment more healthy. Some of these policy changes would be immediate and easy (e.g., changing the tax structure to make good foods cheaper and junk food more expensive, or regulating the removal of the most toxic components of junk food). Other policy changes will take generations - even if we require urban planning to take into account healthy lifestyle promotion, the turnover in infrastructure is so slow that it will take a long time to occur. We certainly need major medical advances, which hopefully our study will aid, because the social changes needed will take decades to fully implement. But that is no reason not to start the public policy debate now, and even the small first steps will save the lives of millions (as well as saving billions from health care budgets).
Nature Genetics interview
Nature Genetics has an interview with me on their blog:
The discovery that NODk mice with the insHEL transgene develop diabetes is described as being serendipitous. What were your initial thoughts about this?
At the time we first found that NODk.insHEL male mice developed diabetes I was actually working on immune defects in NOD mice, rather than beta cell defects. My first thoughts were that this was just another immune defect, with the immune system attacking the beta cells because they expressed the insHEL transgene. Since it fit our preconceived ideas we didn’t take too much notice, but just to be safe I set up a backcross to eradicate the adaptive immune system from the NODk.insHEL mice. It took a couple of years for the mice to breed and age, so I had almost forgotten about the finding when the first immune-deficient NODk.insHEL mice started to develop diabetes. At that point I was really startled – the cross should have eliminated diabetes if it was immune-mediated. I knew then that we were looking at some completely new biology – which took another 10 years to dissect!
What advantages does your new mouse model bring to the field?
There are so many aspects to diabetes that it is often impossible to untangle the causes of disease. For example, one of the critical clinical developments in type 2 diabetes is the death of beta cells. It marks a shift from insulin-resistant diabetes (which is largely treatable), to insulin-deficient/insulin-resistant diabetes (for which there are no effective treatments). But why are the beta cells dying? From previously mouse models there were many reasonable hypotheses that were put forward – maybe it is the demand placed on the beta cell for extra insulin production, maybe it is a toxic effect of high blood glucose levels, maybe it is a side-effect of the high fat diet used to induce diabetes in the first place, or maybe it is immune-mediated. Our model has the advantage that it can strip away all of these interactions to observe the direct effects of forcing beta cells to produce too much protein – a process that results in beta cell failure. Looking forward, I see a major advantage in using this model to screen for drugs that stop the loss of beta cells in type 2 diabetes, which is really the key unmet medical need in diabetes treatment.
One of your interesting observations is the difference in diabetes incidence between the male and female mice, mediated by male sex hormones. What parallels are there with humans and how might you use this model to explore this further?
So far we have only seen diabetes in male insHEL mice, despite challenging female insHEL mice with multiple strategies that promote diabetes in male mice (diet, autoimmunity, genetic background). At a cellular level it looks like male islets are just under more metabolic pressure than female islets, such that the insHEL stress is enough to make male mice diabetic, while female mice stay healthy. This could actually explain a lot about the epidemiology of type 1 diabetes in humans. Most autoimmune diseases have a strong female bias, while type 1 diabetes has a weak male bias. Our hypothesis is that maybe males have an intrinsic islet fragility (perhaps from supporting a larger body mass), while females have an intrinsic susceptibility to autoimmune disease. In epidemiological terms, these two effects may cancel each other out, leading to similar levels of type 1 diabetes in males and females, but at a clinical level it may mean that different individuals would respond better to different treatment strategies.
You identified two loci linked with insHEL-induced diabetes in the NODk mice. What were your expectations about what you would find? Where you excited when Xrcc4 and Glis3 were identified as candidate genes?
Geneticists have been trying to work out the basis of spontaneous diabetes in NOD mice since the strain was first published in 1980. It turns out to be a very complex problem – there are more than 20 loci that contribute to diabetes susceptibility, and each time a loci is analysed in detail it ends up being a cluster of weaker loci working together. Decades later and we are only sure about a handful of candidates genes – so I didn’t have high expectations that we would progress far when looking at the genetics of insHEL-triggered diabetes. It turns out, however, that we had several major advantages. First, the genetics ended up being much simpler, with linkage only observed on two chromosomes. Second, because we knew which cell type was important – the beta cell – we were not operating in the dark about candidates. After filtering for expression in beta cells we were left with only a handful of candidates. Seeing Xrcc4 and Glis3 on the final list was bliss – they both made perfect biological sense. GLIS3 is one of the very few genes linked to both type 1 and type 2 diabetes in humans, and here we had it on our shortlist for a model that contains aspects of both diseases! It had taken more than 10 years to get to those two genes, but then we reached one of those dream runs in the laboratory where all the data just comes together, and every experiment gave support to the candidates.
You identify beta cell failure as a common link between T1D and T2D. Are there ways that your findings can impact the clinical understanding or management of these diseases?
In some ways, what we have here is the laboratory catching up to the clinic. The clinical overlaps between type 1 and type 2 diabetes have been apparent from the start, yet the research on genetics and animal models has consistently emphasized the differences. We may be in the process of reconciling these two approaches. The model that I favour is one where beta cell robustness or fragility lies at the centre of both diseases. In type 1 diabetes, failures in immune tolerance promote an attack on the beta cell, while in type 2 diabetes, hepatic insulin resistance leads to beta cell stress. In both cases, however, it may be the intrinsic robustness or fragility of the beta cell that dictate whether the pressure on beta cells remains subclinical or leads to diabetes. If this model holds true in patients then it would present a golden opportunity for preventing diabetes by increasing the robustness of beta cells.
New study may lead to improved treatment of type 2 diabetes
Genetic cause found for loss of beta cells during diabetes development
Worldwide, 400 million people live with diabetes, with rapid increases projected. Patients with diabetes mostly fall into one of two categories, type 1 diabetics, triggered by autoimmunity at a young age, and type 2 diabetics, caused by metabolic dysfunction of the liver. Despite being labeled a “lifestyle disease”, diabetes has a strong genetic basis. New research under the direction of Adrian Liston (VIB/KU Leuven) has discovered that a common genetic defect in beta cells may underlie both forms of diabetes. This research was published in the international scientific journal Nature Genetics.
Adrian Liston (VIB/University of Leuven): “Our research finds that genetics is critical for the survival of beta cells in the pancreas – the cells that make insulin. Thanks to our genetic make-up, some of us have beta cells that are tough and robust, while others have beta cells that are fragile and can’t handle stress. It is these people who develop diabetes, either type 1 or type 2, while others with tougher beta cells will remain healthy even in if they suffer from autoimmunity or metabolic dysfunction of the liver.”
Different pathways to diabetes development
Diabetes is a hidden killer. One out of every 11 adults is suffering from the disease, yet half of them have not even been diagnosed. Diabetes is caused by the inability of the body to lower blood glucose, a process normally driven by insulin. In patients with type 1 diabetes (T1D), this is caused by the immune system killing off the beta cells that produce insulin. In patients with type 2 diabetes (T2D), a metabolic dysfunction prevents insulin from working on the liver. In both cases, left untreated, the extra glucose in the blood can cause blindness, cardiovascular disease, diabetic nephropathy, diabetic neuropathy and death.
In this study, an international team of researchers investigated how genetic variation controls the development of diabetes. While most previous work has focused on the effect of genetics in altering the immune system (in T1D) and metabolic dysfunction of the liver (in T2D), this research found that genetics also affected the beta cells that produce insulin. Mice with fragile beta cells that were poor at repairing DNA damage would rapidly develop diabetes when those beta cells were challenged by cellular stress. Other mice, with robust beta cells that were good at repairing DNA damage, were able to stay non-diabetic for life, even when those islets were placed under severe cellular stress. The same pathways for beta cell survival and DNA damage repair were also found to be altered in diabetic patient samples, indicating that a genetic predisposition for fragile beta cells may underlie who develops diabetes.
Adrian Liston (VIB/University of Leuven): “While genetics are really the most important factor for developing diabetes, our food environment can also play a deciding role. Even mice with genetically superior beta cells ended up as diabetic when we increased the fat in their diet.”
A new model for testing type 2 diabetes treatments
Current treatments for T2D rely on improving the metabolic response of the liver to insulin. These antidiabetic drugs, in conjunction with lifestyle interventions, can control the early stages of T2D by allowing insulin to function on the liver again. However during the late stages of T2D, the death of beta cells means that there is no longer any insulin being produced in the pancreas. At this stage, antidiabetic drugs and lifestyle interventions have poor efficacy, and medical complications arise.
Dr Lydia Makaroff (International Diabetes Federation): “The health cost for diabetes currently exceeds US$600 billion, 12% of the global health budget, and will only increase as diabetes becomes more common. Much of this health care burden is caused by late-stage type 2 diabetes, where we do not have effective treatments, so we desperately need new research into novel therapeutic approaches. This discovery dramatically improves our understanding of type 2 diabetes, which will enable the design of better strategies and medications for diabetes in the future”.
Adrian Liston (VIB/University of Leuven): “The big problem in developing drugs for late-stage T2D is that, until now, there has not been an animal model for the beta cell death stage. Previously, animal models were all based on the early stage of metabolic dysfunction in the liver, which has allowed the development of good drugs for treating early-stage T2D. This new mouse model will allow us, for the first time, to test new antidiabetic drugs that focus on preserving beta cells. There are many promising drugs under development at life sciences companies that have just been waiting for a usable animal model. Who knows, there may even be useful compounds hidden away in alternative or traditional medicines that could be found through a good testing program. If a drug is found that stops late-stage diabetes, it would really be a major medical breakthrough!”
Read more: Dooley*, Tian*, Schonefeldt*, Delghingaro-Augusto*, Garcia-Perez, Pasciuto, Di Marino, Carr,Oskolkov, Lyssenko, Franckaert, Lagou, Overbergh, Vandenbussche, Allemeersch, Chabot-Roy, Dahlstrom, Laybutt, Petrovsky, Socha, Gevaert, Jetten, Lambrechts, Linterman, Goodnow, Nolan, Lesage, Schlenner**, Liston**. 'Genetic predisposition for beta cell fragility underlies type 1 and type 2 diabetes.' Nat Genet. 2016
There is nothing ethical about a moratorium on genome editing
In recent weeks, two comment pieces have been published calling for a moratorium on germ-line genome editing. Germ-line genome editing is now feasible, even easy, thanks to new genetic engineering tools such as CrispR-Cas9. It has the potential to fix genetic diseases such as Huntington's, severe combined immunodeficiency and cystic fibrosis. So why is there suddenly a call for a moratorium on curing these diseases? Are there new scientific results questioning the safety or efficacy? No. In fact new studies are making the approach more and more realistic every day. This moratorium call is just a commentary, not based on any new science. The only reason it made headlines is that the two commentaries were published in the leading journals, one in Science and one in Nature, and because the 23 authors include some very preeminent scientists (mandatory to have your comment pieces published in Science and Nature). This was widely reported as "Scientists seek ban on editing human genome", and I expect that various bans will indeed soon be implemented across the world.
My question - as a scientist who works on genome editing using these very tools - is why? Why is there a call for a ban? The case has simply not been made that there is any ethical conundrum. The "problem" is that these cures would not only cure the disease in the child, but would also prevent the disease being passed on to their children. And why exactly is that a bad thing? "Future generations" - yadda yadda. We make decisions that influence future generations every single day. Do you think future generations will complain about not having cystic fibrosis? If need be, they could easily engineer the mutation back in, not that anyone would. You know what else potentially causes germ-line mutations? X-rays, but we don't ban them because the benefits are very large and the risks are very small. Even poverty causes heritable modification to the genome, so let's not pretend that we've never made a decision that alters unborn generations.
To me, all this moralising is more of the same that we have heard for decades about "designer babies". I'm sick and tired of hearing about the hypothetical of chosing a baby's eye colour. That is probably never going to happen, it would be easy to ban if it did start to happen, and is it really any worse than the current practise of grooming children for beauty pagents or gymnastics from an early age?
The hypotheticals that bioethicists seem to be overwhelmed about always seem to be in the indefinite future. So I'd like to give a here-and-now question to the authors of those comment pieces, and to bioethicists in general. Is it ethical to withhold medicine to children today, simply because of some ill-defined unease you have? The picture below is of a child with Olmsted disease, which we work on in the lab. Warning: the picture is not nice, but this is exactly the type of disease which could be potentially cured with the new genome editing tools.
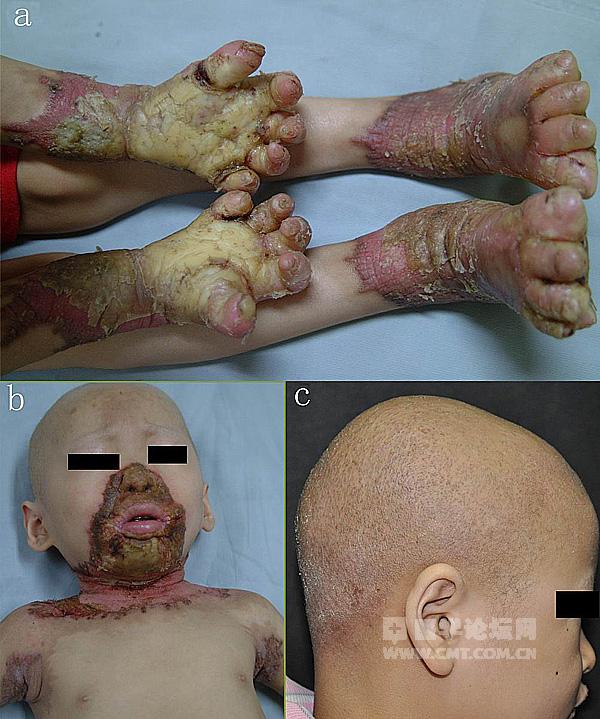
Olmsted disease is caused by a single base-pair mutation in the gene TRPV3. It is a prime candidate for genome editing cures, but any cure would run the "risk" of not just correcting the mutation in the skin, but also of correcting the mutation in the germ-line. Is it ethical to cure such a child at the "risk" of also curing their future children? I would argue that not only is it ethical, but it is unethical to not try.
There are horrible diseases which strike down children that may never have any feasible cure other than genome editing. To not pursue that sole avenue of research would be a disgrace, an ethical failure of the highest magnitude. I, for one, will ignore any self-proclaimed "moral authority" who tells me not to work for a cure of these diseases. Unless my research is proclaimed illegal I will continue my work - and if it is proclaimed illegal I'll campaign against the unethical laws that shut down the sole hope of families with incurable genetic diseases. Ethical action requires a careful consideration of the consequences, but equally, inaction also requires a a consideration of the ethical consequences. Unless a strong case is made that the consequences of genome-editing for future generations are worse than the consequences of not using genome-editing for this generation, it would be unethical to abide by a moratorium.
New cause for early-onset lupus discovered
In a new study out by the Autoimmune Genetics Laboratory, we have discovered a new genetic cause for early-onset systemic lupus erythematosus - mutation in the gene IFIH1. In 2014, mutations of this gene were independently found to cause the neurodegenerative disease Aicardi-Goutières syndrome (AGS). Despite lupus and AGS manifesting as clinically different symptoms, this study shows that mutation in the same gene causes both diseases. The mutation in IFIH1 works via driving excessive production of the cytokine IFN alpha, so this discovery opens up the possibility for treatment once anti-IFN alpha antibodies (currently in development) are approved for use.

Read more: Van Eyck, De Somer, Pombal, Bornschein, Frans, Humblet-Baron, Moens, de Zegher, Bossuyt, Wouters* & Liston*. IFIH1 mutation causes systemic lupus erythematosus with selective IgA-deficiency. Arthritis Rheumatol. 2015, in press.
If you would like to support our clinical research, and allow us to take on more cases like this one, you can make a tax-deductable donation the Ped IMID fund, by transferring to IBAN-number BE45 7340 1941 7789, BIC-code: KREDBEBB with the label "voor EBD-FOPIIA-O2010".
New fund to support translational research into paediatric inflammatory diseases
A new fund has been set up to drive bench-to-bedside research for children with inflammatory immune diseases. The Ped IMID fund (Fonds Pediatrische Immuun-inflammatoire Aandoeningen) was set up by Prof Carine Wouters (Pediatric Rheumatology), Prof Patrick Matthys (Immunobiology) and Prof Adrian Liston (Autoimmune Genetics) to build on our strong research cooperation. More than merely "translational research", where basic science is pushed into the clinic, our group performs "dialog research", where we meet regularly to discuss the clinic and the science of the most difficult-to-treat patients. We use the clinic to inform the research and the research to inform the clinic, and have already had multiple break-throughs in understanding and treating children with rare inflammatory diseases.

If you would like to support our research, and allow us to take on more cases, you can transfer a tax-deductable donation to IBAN-number BE45 7340 1941 7789, BIC-code: KREDBEBB with the label "voor EBD-FOPIIA-O2010".

New disease (and cure!) found
As part of an ERC funded research program, the Autoimmune Genetics Laboratory is searching the genomes of young children with severe immune diseases to look for novel genes (and hopefully treatments). In a collaboration with Prof Carine Wouters and Prof Isabelle Meyts at UZ Leuven, we found mutations in a new gene, CECR1, in three severely ill children. Two of the children were born with a severe immune deficiency, making them prone to infections, while the third developed an inflammatory disease known as Castleman's disease. Mutations in the same gene, which produces the protein ADA2, were independently found by two other groups to give vascular disease and early-onset stroke.
These studies identify ADA2-deficiency as a previously undiagnosed primary immunodeficiency which includes components of immune deficiency, inflammation and vasculopathy. Most importantly, this new diagnosis comes with a successful cure: prior to genetic diagnosis, our clinical collaborators were able to successfully treat the disease with bone-marrow transplanation (for the immunodeficient patient) or tocilizumab (for the Castleman's disease patient). These results therefore not only add to our knowledge about medical genetics, but also provide a direct diagnosis-treatment pathway for any new children identified with these severe diseases.

Read more:
Van Eyck, Hershfield, Pombal, Kelly, Ganson, Moens, Frans, Schaballie, De Hertogh, Dooley, Bossuyt, Wouters, Liston* and Meyts*. Hematopoietic stem cell transplantation rescues the immunologic phenotype and prevents vasculopathy in patients with adenosine deaminase 2 deficiency. J Allergy Clin Immunol. 2015 Jan;135(1):283-287.e5.
Van Eyck, Liston and Wouters. Mutant ADA2 in vasculopathies. N Engl J Med. 2014 Jul 31;371(5):480
Van Eyck, Liston and Meyts. Mutant ADA2 in vasculopathies. N Engl J Med. 2014 Jul 31;371(5):478-9.
If you would like to support our clinical research, and allow us to take on more cases like this one, you can make a tax-deductable donation the Ped IMID fund, by transferring to IBAN-number BE45 7340 1941 7789, BIC-code: KREDBEBB with the label "voor EBD-FOPIIA-O2010".